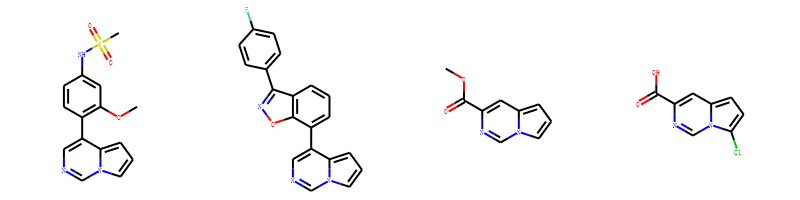
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
| from rdkit.Chem import Draw
from rdkit import Chem
smis=[
'COC1=C(C=CC(=C1)NS(=O)(=O)C)C2=CN=CN3C2=CC=C3',
# 'CCN(CC1=C(C=CC(=C1)C(F)(F)F)C2=CC(=C3N2C=NC=C3)CC(=O)O)C(=O)C4CC4',
'C1=CC2=C(C(=C1)C3=CN=CN4C3=CC=C4)ON=C2C5=CC=C(C=C5)F',
'COC(=O)C1=CC2=CC=CN2C=N1',
'C1=C2C=C(N=CN2C(=C1)Cl)C(=O)O',
]
template = Chem.MolFromSmiles('c1nccc2n1ccc2')
AllChem.Compute2DCoords(template)
mols=[]
for smi in smis:
mol = Chem.MolFromSmiles(smi)
AllChem.GenerateDepictionMatching2DStructure(mol,template)
mols.append(mol)
img=Draw.MolsToGridImage(mols,molsPerRow=4,subImgSize=(200,200),legends=['' for x in mols])
img
|